








Azot, podobnie jak wiele innych pierwiastków chemicznych, posiada zdolność do katenacji. Różnorodność i wielość struktur molekularnych zawierających wiązanie azot-azot jest istotnie mniejsza niż w przypadku węgla.
Azot, podobnie jak wiele innych pierwiastków chemicznych, posiada zdolność do katenacji. Różnorodność i wielość struktur molekularnych zawierających wiązanie azot-azot jest istotnie mniejsza niż w przypadku węgla. Związki takie cieszą się szczególnym zainteresowaniem jako potencjalne materiały wysokiej gęstości energii (HEDM - high-energy density materials) [1], gdyż mogą one ulegać reakcji egzotermicznej Nx → (x/2)N2 z wydzieleniem 50 kcal/mol i więcej w przeliczeniu na jeden atom azotu [2]. Niemniej jednak ich praktyczne zastosowania ogranicza ich stabilność. Znane są zarówno układy łańcuchowe, cykliczne jak i klatkowe zawierające wiązanie azot-azot. Badania eksperymentalne wykazały, iż możliwe jest otrzymanie jonów N3 − do N5 + w warunkach laboratoryjnych [3÷5] jak również sieci polimerowych [6] ale tylko pod wpływem wysokiego ciśnienia (110 GPa). Na podstawie zasobów krystalograficznej bazy danych Cambridge Structural Database (CSD) [7], można stwierdzić, iż największą grupę związków chemicznych stanowią układy zawierające dwa związane ze sobą atomy azotu (ok. 85% wszystkich struktur typu Nx, gdzie x≥2). Wśród tych układów najliczniejszą grupę stanowią -hydrazo, -azo, -azoksy, -azodioksy oraz związki zawierające grupę nitraminową. Cztery pierwsze stanowią względem siebie szereg utleniająco-redukujący. Na grupę funkcyjną hydrazo składają się dwa atomy azotu sp3 połączone wiązaniem ó oraz dodatkowo każdy z nich tworzy wiązanie chemiczne z atomem wodoru. Natomiast dwa atomy azotu o hybrydyzacji sp2, z których każdy posiada wolną niewiążącą parę elektronową, połączone ze sobą wiązaniem podwójnym tworzą dwuwartościową grupę azową. Cząsteczki związków chemicznych zawierające tę grupę funkcyjną mogą występować jako dwa izomery geometryczne E i Z. W wyniku utworzenia wiązania chemicznego atomu tlenu z wolną parą elektronową, pochodzącą od jednego z atomów azotu grupy azowej, powstaje grupa funkcyjna azoksy. Azoksyzwiązki, nazywane N-tlenkami diazenów, również wykazują izomerię geometryczną Z i E względem wiązania azot-azot. Gdy oba atomy azotu grupy azowej utworzą wiązanie chemiczne z atomem tlenu (>N2O2), wtedy powstałą grupę przyjęto nazywać jako azodioksy. Stanowi ona najbardziej utlenioną postać wiązania azot-azot [8÷14]. Inny porządek powiązania atomów tlenu i azotu w obrębie grupy >N2O2 (N-NO2) spotykany jest w klasie związków nazywanych nitraminami.
Grupa nitraminowa tworzy w tych związkach czterordzeniowy układ ð-elektronowy, który może oddziaływać z podstawnikami amidowego atomu azotu. Badania właściwości wiązania N-N oraz wpływu grupy nitraminowej na aromatyczność pierścienia w grupie fenylowych pochodnych nitramin podyktowane są również chęcią wyjaśnienia mechanizmu tzw. przegrupowania nitraminowego.
Opis
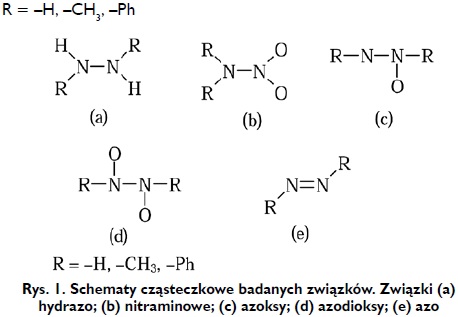
Do analizy strukturalnej i elektronowej grup funkcyjnych zawierających wiązanie azot-azot wybrano układy pokazane na Rysunku 1.
W celu ustalenia zmian w długości wiązania azot-azot w układach pokazanych na Rysunku 1, wyliczono jego średnie wartości na podstawie danych z bazy CSD oraz wyznaczono jego wartości za pomocą obliczeń kwantowo-mechanicznych. Obliczenia te przeprowadzono metodą MP2 [15] stosując bazę funkcyjną 6-311++G(d,p) programem Gaussian09 [16] poprzez pełną optymalizację bez nakładania więzów na geometrię cząsteczki, tj. długości wiązań, wartości kątów walencyjnych czy torsyjnych.
Narzędziem pozwalającym na głębsze "spojrzenie" w strukturę elektronową molekuły jest topologiczna analiza rozkładu gęstości elektronowej oparta na teorii Atoms in Molecules (AIM) [17÷19].
Teoria ta, na podstawie trójwymiarowej funkcji gęstości elektronowej (ń), definiuje podstawowe pojęcia chemiczne, takie jak: atom, wiązanie chemiczne, oddziaływania niewiążące. Zaletą teorii AIM jest jej uniwersalność. Funkcja gęstości elektronowej w cząsteczkach może być analizowana na podstawie zarówno eksperymentu dyfrakcyjnego jak i metod kwantowo-mechanicznych. W analizie największe zastosowanie posiadają dwa z czterech punktów stacjonarnych funkcji gęstości elektronowej; punkt krytyczny wewnątrz pierścienia (z ang. Ring Critical Point, RCB) oraz punkt krytyczny wiązania (z ang. Bond Critical Point, BCP), znajdujący się między dwoma sąsiadującymi jądrami atomowymi. Punkty krytyczne odpowiadają maksimum, punktom siodłowym oraz lokalnemu minimum funkcji gęstości elektronowej i wyznaczane są na podstawie topologii rozkładu gęstości elektronowej. Aby właściwie je scharakteryzować należy obliczyć Hesjan, czyli macierz drugich pochodnych funkcji gęstości po wszystkich możliwych kombinacjach współrzędnych. Diagonalizacja takiej macierzy prowadzi do uzyskania trzech wartości własnych Hesjanu, których ranga (rank, liczba niezerowych wartości własnych) i wartość (signature, suma znaków niezerowych wartości własnych Hesjanu) charakteryzują punkty krtytyczne [19]. Dla RCP własności te przyjmują postać (3,+1), natomiast dla BCP (3,-1). Wartość gęstości elektronowej w tych punktach jest miarą ilości ładunku elektronowego.
Wiązania chemiczne wykazujące charakter bardziej kowalencyjny posiadają zwykle większą wartość gęstości elektronowej w punkcie krytycznym niż wiązania o charakterze jonowym.
Układy, w których wiązanie azot-azot pełni rolę mostka pomiędzy pierścieniami aromatycznymi, stanowią doskonały model do opisu i analizy efektów elektronowych będących skutkiem ich wzajemnego oddziaływania. W celu wyznaczenia wpływu na aromatyczność pierścieni fenylowych połączonych z analizowanymi grupami funkcyjnymi, na podstawie geometrycznego kryterium aromatyczności, zastosowano indeks HOMA (z ang.Harmonic Oscillator Model of Aromaticity), który zdefiniowany jest równaniem 1.
gdzie n jest liczbą sumowanych wiązań chemicznych; á - stałą normalizacyjną (dla wiązań C-C á = 257,7) ustaloną tak, aby HOMA = 0 dla układu niearomatycznego oraz HOMA = 1 dla układu, w którym wszystkie długości wiązań są równe optymalnej wartości Ropt, zakładającej w pełni aromatyczny układ (dla wiązań C-C, Ropt równe jest 1,388 Å);
Ri - długość i-tego wiązania [20, 21].
W Tablicy 1 zebrano średnie długości wiązań N-N oraz odpowiadające im wartości gęstości elektronowej w badanych grupach związków. Największa długość jest obserwowana w przypadku związków hydrazo, najmniejsza natomiast dla azozwiązków. Wynika to z faktu, iż pomiędzy atomami azotu w układach hydrazo występuje wiązanie pojedyncze, natomiast w związkach azo podwójne.
W przypadku związków azoksy i azodioksy, długość wiązania jest pomiędzy długością wiązania pojedynczego a podwójnego.
W strukturach nitramin średnia odległość między atomami azotu jest o 0,020 Å krótsza niż w przypadku związków hydrazo, jednocześnie jest wyraźnie dłuższa (o 0,147 Å) od średniej wartości dla azozwiązków. Wraz ze skracaniem się tego wiązania następuje wzrost wartości gęstości elektronowej w punktach krytycznych tych wiązań. Otrzymane za pomocą obliczeń kwantowo-mechanicznych długości mają zbliżone wartości do tych otrzymanych z bazy danych CSD [7]. Największą długość wiązania zaobserwowano dla cząsteczki niepodstawionej nitraminy, która wynosi 1,460 Å. Natomiast najkrótsze (1,373 Å) dla N-fenylonitraminy.
Różnica ta wskazuje na znaczący wpływ podstawników amidowego atomu azotu na strukturę grupy nitraminowej (wiązanie N-N).
Co więcej, również wzajemne ułożenie podstawnika w stosunku do płaszczyzny grupy nitrowej ma wpływ na długość omawianego wiązania. Obserwowane jest to dla cząsteczki N-fenylonitraminy, w której w zależności od orientacji (prostopadłej/równoległej) długość zmniejsza się o 0,041 Å, w przypadku, gdy grupa nitaminowa jest koplanarna z pierścieniem aromatycznym. Wpływ na taki stan rzeczy może mieć fakt, iż dwa ð-elektronowe systemy w takim ułożeniu mogą ze sobą oddziaływać, tworząc dziesięcio-rdzeniowy układ zdelokalizowanych elektronów.
W Tablicy 2 zebrano wartości indeksu HOMA dla pierścieni fenylowych związanych z analizowanymi grupami funkcyjnymi zawierającymi wiązanie azot-azot. Indeks aromatyczności HOMA jest taki sam w przypadku związków hydrazo i azo. Wskazuje to na identyczny wpływ tych grup na aromatyczność pierścieni fenylowych, którymi są podstawione oraz brak udziału wiązania ð w delokalizacji na mostku N-N w przypadku związków azowych.
Indeks HOMA dla pierścieni fenylowych azoksyzwiązków wynosi 0,970 dla pierścienia związanego z utlenionym atomem azotu, oraz 0,911 dla pierścienia połączonego z drugim atomem azotu. Różnica ta może być efektem zjawiska rezonansu zachodzącego w obrębie cząsteczki. Wynika z niego, iż pierścień oddalony od atomu tlenu grupy N-N może tracić na aromatyczności ze względu na tworzącą się strukture chinoidową (Rys. 2).
Wartość indeksu aromatyczności w przypadku pierścienia fenylowego związanego z utlenionym atomem azotu jest taka sama jak w przypadku związków azodioksy (0,970).
Warto w tym miejscu zwrócić uwagę na asymetrię oddziaływania mostka azoksy z pierścieniami fenylowymi. W analizowanych układach, największe zmiany w aromatyczności obserwuje się dla pierścienia związanego z nieutlenionym atomem azotu, podczas gdy najmniejsze, w przypadku gdy pierścień związany jest z utlenionym atomem azotu mostka azoksy.
Analizując wartości indeksu aromatyczności HOMA w nitraminach aromatycznych odnajdujemy potwierdzenie powstania skoniugowanego układu C6H5NNO2. Indeks ten dla wszystkich badanych cząsteczek nitramin oscyluje w granicach 0,962 - 0,966, natomiast w przypadku płaskiej cząsteczki N-fenylonitraminy, jego wartość zmniejsza się do 0,952 (dla ułożenia prostopadłego HOMA = 0,964).
Podsumowanie
Do analizy strukturalnej i elektronowej grup funkcyjnych zawierających wiązanie azot-azot wybrano związki chemiczne zawierające grupy: hydrazo; azo; azoksy; azodioksy oraz nitraminową.
W celu ustalenia zmian w długości wiązania azot-azot wyliczono jej średnie wartości na podstawie obliczeń kwantowo-mechanicznych oraz danych z bazy CSD [7]. Dodatkowo wyznaczono wpływ analizowanych grup funkcyjnych na aromatyczność związanych z nimi pierścieni fenylowych, poprzez wyliczenie indeksu aromatyczności HOMA.
Największa wartość długości wiązania azot-azot jest obserwowana w przypadku związków hydrazo, najmniejsza natomiast dla azo-związków. Pozostałe grupy charakteryzują się wartościami pośrednimi.
Obserwowana jest duża korelacja pomiędzy wartościami gęstości elektronowej w punkcie krytycznym wiązania azot-azot a jego długością. Indeks aromatyczności HOMA zwiększa się w szeregu związków od azoksy poprzez azo/hydrazo, nitraminy do azodioksy, wskazując na malejący wpływ tych grup na delokalizację ð-elektronową związanych z nimi pierścieni fenylowych. Ponadto w grupie azoksy zaobserwowano zróżnicowanie w oddziaływaniu z układami aromatycznymi oraz w przypadku nitramin wykazano wpływ wzajemnego położenia względem siebie pierścienia oraz związanej z nim grupy funkcyjnej.
Podziękowania
Autorzy dziękują Wrocławskiemu Centrum Sieciowo-Superkomputerowemu za umożliwienie wykonania obliczeń kwantowo-mechanicznych. Katarzyna Gajda jest stypendystką projektu systemowego Samorządu Województwa Opolskiego pn. "Stypendia doktoranckie - inwestycja w kadrę naukową województwa opolskiego" na rok akademicki 2013/14.
Literatura
1. Strout D. L.: Why Isn’t the N20 Dodecahedron Ideal for Three-Coordinate Nitrogen?. J. Phys. Chem. A 2005, 109, 1478-1480.
2. Fau S., Bartlett R. J.: Possible products of the end-on-addition of N3 − to N5 +. J. Phys. Chem. A 2001, 105, 4096-4106.
3. Christe K. O., Wilson W. W., Sheehy J. A.; Boatz J. A.: N5 +: A Novel Homoleptic Polynitrogen Ion as a High Energy Density Material. Angew. Chem., Int. Ed. 1999, 38, 2004-2009.
4. Vij A., Pavlovich J. G., Wilson W. W., Vij V., Christe K. O.: Experimental Detection of the Pentaazacyclopentadienide (Pentazolate) Anion, cyclo- N5 − Angew. Chem., Int. Ed. 2002, 41, 3051-3054.
5. Butler R. N., Stephens J. C., Burke L. A.: First generation of pentazole (HN5, pentazolic acid), the final azole, and a zinc pentazolate salt in solution: A new N-dearylation of 1-(p-methoxyphenyl) pyrazoles, a 2-(p-methoxyphenyl) tetrazole and application of the methodology to 1-(p-methoxyphenyl) pentazole. Chem. Commun. 2003, 8, 1016-1017.
6. Eremets M. I., Gavriliuk A. G., Trojan I. A., Dzivenko D. A. Boehler R.: Single-bonded cubic form of nitrogen. Nature Mater. 2004, 3, 558-563.
7. Allen F. H.: The Cambridge Structural Database: a quarter of a milion crystal structures and rising. Acta Cryst. 2002, B58, 380-388.
8. Shorter J.: The chemistry of hydrazo-, azo- and azoxy- groups S. Patai, Ed., J. Wiley, N. York, 1997, 2, 367.
9. Kyzioł J.B., Ejsmont. K.: 9-Methyl-3-phenyldiazenyl-9H-carbazole; X-ray and DFT calculated structures. Acta. Cryst. 2007, C63, 77-79.
10. Ejsmont K., Domański A.A., Kyzioł J.B., Zaleski J.: Disorder in the crystals of trans-4-fluoroazoxybenzene. Synthesis, Spectral Properties, Crystal Structures and DFT Calculations J. Mol. Struct., 2005, 753, 92-98.
11. Ejsmont K., Domański A., Kyzioł J.B., Zaleski J.: 4-Hydroxy-ONN-azoxybenzene Acta Cryst., 2000, C56, 697-699.
12. Domański A., Ejsmont K., Kyzioł J.B., Zaleski J.: Two trans-4-aminoazoxybenzenes. Acta Cryst., 2001, C57, 467-470.
13. Ejsmont K., Broda M., Domański A.A., Kyzioł J.B., Zaleski J.: Orthorhombic polymorphs of two trans-4-aminoazoxybenzenes. Acta Cryst., 2002, C58, 545-548.
14. Ejsmont K., Domański A.A., Kyzioł J.B., Zaleski J.: Trans-4-bromo-ONN- azoxybenzene at 100 K. Acta Cryst., 2004, C60, 368-370.
15. Møller C., Plesset M.: Note on an approximation treatment for manyelectron systems. Phys. Rev., 1934, 46, 618-622.
16. Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
17. Bader R. F. W. Atoms in Molecules: A Quantum Theory, Clarendon, Oxford, 1990.
18. Ronald J. Gillespie and Paul L. A. Popelier, Chemical Bonding and Molecular Geometry: From Lewis to Electron Densities, Oxford University Press: New York, NY, 2001.
19. Matta C. F. and Boyd R. J. An Introduction to the Quantum Theory of Atoms in Molecules, in The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007.
20. Krygowski T. M.: Crystallographic studies of inter- and intramolecular interactions reflected In aromatic character of ð-electron system J. Chem. Inf. Comput. Sci., 1993, 33, 70-78.
21. Krygowski T. M.; Cyrański M. K.: Separation of the energetic and geometric contributions to the aromaticity. Part IV. A general model for the ð-electron systems. Tetrahedron 1996, 52, 10255-10264.












KOMENTARZE (0)
Do artykułu: Strukturalne i elektronowe aspekty wiązania azot-azot